

Para-substituted aromatic compounds are an excellent way to explore electronic effects in NMR spectroscopy. In this comparison, we will look at 1,4-diethylbenzene and 4-nitrotoluene. The two compounds share a similar para-substituted benzene framework, but their substituents affect the ring's electron density in very different ways. The ethyl groups in 1,4-diethylbenzene are weak electron donors, they increase electron density through inductive effects and hyperconjugation. This added electron density shields the aromatic protons, causing their resonances to appear upfield between 7.0 - 7.25 ppm. In contrast, the nitro group in 4-nitrotoluene is one of the strongest electron-withdrawing substituents commonly encountered in aromatic chemistry. Through both inductive and resonance effects, the nitro group pulls electron density away from the ring, reducing shielding around nearby protons and shifting their resonances downfield between 7.27 - 8.25 ppm.

125 MHz ¹H NMR spectra of 1,4-diethylbenzene and 4-nitrotoluene acquired on the QM-125 with both samples at 200 mM in CDCl₃ using 256 scans, a 2 s acquisition time, and 6 s recovery delay.
The aromatic resonances of 4-nitrotoluene appear noticeably downfield relative to those of 1,4-diethylbenzene because the protons experience a more electron-deficient environment. Since both molecules are para-substituted, their spectra remain relatively simple and easy to interpret, allowing the influence of substituent electronics to stand out without the added complexity of asymmetric substitution patterns. This makes the pair an excellent teaching example for demonstrating the relationship between molecular structure and NMR chemical shifts.
On the QM-125 125 MHz benchtop NMR, these trends are readily observed. The QM-125 delivers the resolution and sensitivity needed to clearly distinguish these electronic effects, bringing structural insights directly to the benchtop without the complexity of a traditional high-field NMR system.

If you have ever wanted the ability to run and automate NMR directly in your own lab or follow a reaction as it unfolds, check out our application note which demonstrates use of the QM-125 to monitor a Fischer esterification reaction. Using QM Control together with a short Python script, the instrument automatically collected spectra every five minutes during a three-hour Fischer esterification of benzoic acid, all performed in methanol. The combination of automated sampling and the built-in flow path of the QM-125 allows the reaction to be viewed step by step through changes in peak integrals for the starting material, intermediate species, and final product. In this reaction, we spot the ortho-ester reaction intermediate as the reaction progresses towards the final product.
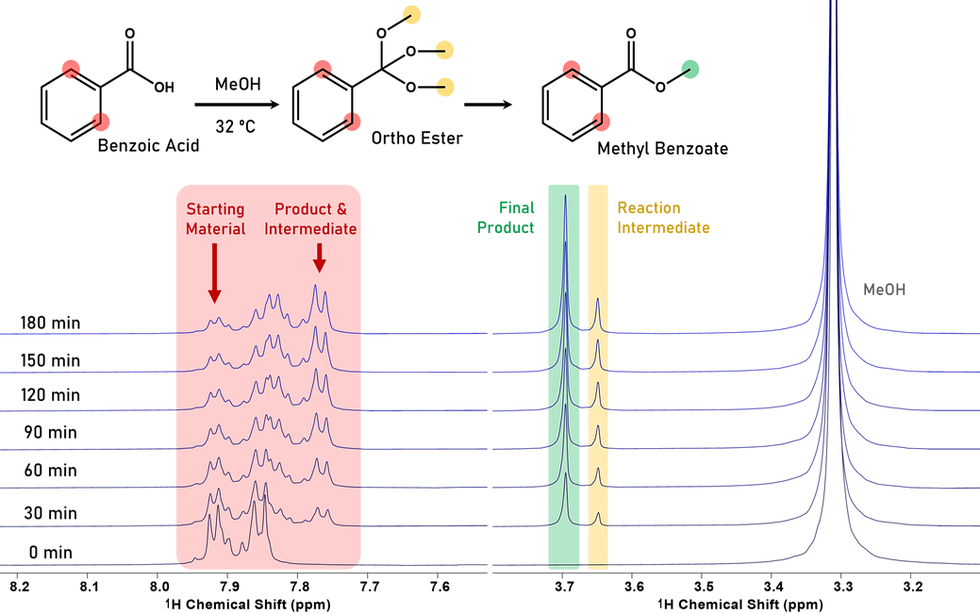
Tracking the progress of a Fischer esterification reaction of benzoic acid in methanol by benchtop NMR at 125 MHz over the course of three hours.
The same flow-ready design makes the QM-125 ready to handle a wide range of customized and hyphenated setups. Whether your workflow is straightforward or complex, it can be automated to collect data at any time-intervals defined by the user. In addition, the QM-125 does not require deuterated solvents and provides consistent field uniformity, which is desired when comparing data at various time points. The application note offers an use case of how automated NMR can be added to modern flow chemistry, process optimization, and other research environments that depend on steady, informative analytical feedback. Most importantly, it shows how chemists can bring NMR directly into their own workflow and let the instrument adapt to their chemistry.
Click the link above to get this application note and learn more about how the QM-125 can be integrated into your lab.
Updated: Oct 27, 2025
The QM-125 benchtop NMR represents a new generation of analytical instrumentation built around flexibility, automation, and simplicity. With its 3-Tesla magnet and 125 MHz Larmor frequency, it delivers the highest field strength in its class while introducing a versatile approach to NMR sample handling: sample-delivery-by-flow. NMR samples can be introduced by syringe and from on-line and in-line automated delivery systems. The versatility of flow-NMR can change the way you think about routine sample handling and high-throughput NMR applications.
Inside the QM-125, a single piece of 100% PFA polymer tubing runs continuously through the magnet from one front-panel fluid fitting to the other, forming an easily replaced and chemically inert flow path. Due to the low surface adhesion of the PFA tubing, liquid samples are easily displaced with air or flushed with solvent to clear the tube for the next sample. For high-precision applications, the sample tube may also be easily washed with µL amounts of the following sample as a sacrificial volume to prepare the tube for measurement. Either way, the sample tube can be reused indefinitely without risk of carryover or contamination with best practices.
Sample tube have a long lifetime however, as every chemist knows, experiments don’t always go as planned when pushing the boundaries of chemistry. Every now and then, a challenging sample or unexpected reaction mixture may make its way into the flow path. That’s why the QM-125 is designed with practicality in mind, so that replacing the sample tube is quick, inexpensive, and user-manageable.
To better understand the sample tube design of the QM-125, check out our recent video, which provides a step-by-step look at how the flow path works and demonstrates how easy it is to replace the sample tube in just a few minutes. The video offers a view inside the instrument and shows how the practical and user-friendly design of sample-delivery-by-flow makes the QM-125 ready for walk-up use and for your automated applications.
With its seamless flow-NMR operation, compact footprint, and hyphenation capabilities, benchtop NMR is becoming more accessible to all NMR users. The design of the QM-125 sets a new standard for benchtop NMR flexibility, paving the way for the next generation of automated, connected, and high-performance NMR applications.